![]()
|
|
|
 |

|
|
|
|
||
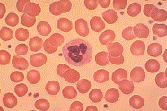
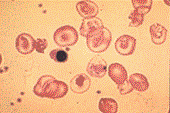
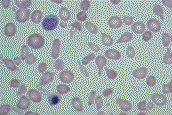
An Overview of Thalassemia for Parents Adopting InternationallyThalassemia refers to a group of inherited anemias (red blood cell deficiency). It is a disease of hemoglobin, which is the protein which delivers oxygen throughout our bodies. It is estimated that 100,000 babies a year are born with severe forms, about 10,000 in India alone. Geographically the thalassemia belt includes the Mediterranean passing through West and Central Asian countries like Turkey, Iran, Afghanistan onto Pakistan & India and passes on to the South East Asian countries like Indonesia, Burma & Thailand, Vietnam and Cambodia. This makes it most common in African, Greek, Italian, Middle Eastern and Southern Asian populations. The two main groups of thalassemia are called alpha and beta. There are four types of alpha thalassemia and three types of beta thalassemia. Alpha thalassemia genes are found in Southeast Asians, blacks, and people originally from the Middle East. The disease affects the red blood cells in two ways. First the body cannot make enough cells with normal hemoglobin. Second, the cells have their lifespan shortened from four months to less than one month. The first type of alpha thalassemia minor (trait) is a carrier state with no anemia and no symptoms. The second type has slightly abnormal red cells but still no anemia. The third type of alpha thalassemias produces a mild anemia that generally doesn't lead to serious complications. The red blood cells are slightly abnormal and resemble iron deficiency anemia. These children would most likely have not been diagnosed and may present for adoption. The fourth type of alpha thalassemia major is the most severe and affects mostly Southeast Asians, Chinese and Filipinos. It results in death before or soon after birth. This last type is usually seen only in Southeast Asians. Beta thalessemias cause a broad range of health symptoms which range from none to very severe. Beta thalassemia genes are more widespread -- found in Africa, the Middle East, India, Southeast Asia, and around the Mediterranean. Someone with only one of the beta genes will have beta thalassemia trait, a mild anemia in which the red cells are smaller than normal. Because small red cells are also typical of iron deficiency anemia, which is extremely common in women and children, many people with this trait are erroneously diagnosed as iron deficient, and are given iron pills. Iron not only doesn't help this mild anemia, but it can build up in the tissues. The three categories of beta thalassemia are major, intermedia and minor. Beta thalassemia minor (thalassemia trait) may cause no symptoms, but can be identified by changes in the blood. Children with thalassemia minor (thalassemia trait) are considered carriers and lead completely normal, healthy lives. Beta thalassemia intermedia is a mild form of Cooley's anemia. Children with thalassemia intermedia may develop similar complications, but usually the disease is mild until adulthood. These children usually do not require transfusions, although they may be recommended if complications start to develop. However, countries such as Vietnam which permit the adoption of very young infants may not have identified these children prior to adoption. Cooley's anemia or thalassemia major is the most severe type. Infants with thalassemia major appear healthy at birth, but develop symptoms around 8 to 10 months of age. These children appear pale, weak, fussy, and have poor appetites. Their growth is slow and they often become jaundiced. Left untreated, the spleen, liver, and heart soon begin to fail. Bones become thin and brittle, and facial bones become misshapen. Untreated children experience retarded physical and mental growth. In untreated children, heart failure and infection are the leading causes of death. Treatment with frequent blood transfusions and antibiotics can alter the course of the illness. Children with thalassemia major can be treated by hypertransfusion. Transfusions every 3 or 4 weeks help keep their hemoglobin near normal. This helps prevent some of the complications of the disease, resulting in better growth and general health and can prevent heart failure and bone deformities in some cases. Unfortunately, the treatments have their own drawbacks. Repeated blood transfusions lead to a buildup of iron in the body, damaging the heart, liver and other organs. This requires daily drug treatment with a chelator to eliminate iron, preventing or delaying problems related to iron overload. The chelator drug is requires daily dosing given by pumping the drug underneath the skin while the child is sleeping. Children treated aggressively with frequent blood transfusions and iron chelation live 20 to 30 years or longer. Bone marrow transplants (BMT) have cured some cases of thalassemia. This high risk treatment is possible only for a small minority of patients as a suitable bone marrow donor must be located. The transplant procedure is still risky and can result in death. The success rate of BMT is as high as 95%, if there is no prior serious organ damage such as excess deposition of iron. About one-third of all BMT patients will have a siblings whose bone marrow is a good match. Adopted children lacking an identifiable common gene pool of their birth family would be less likely to find a suitable donor than a child with an intact and identifiable birth family. Other treatments on the horizon include oral chelating drugs and various types of gene therapy, including stem cell transplants. All thalassemias are inherited. People unfamiliar with the disease may mistakenly think it can be caught from another child. Blood tests can show whether an adopted child has thalassemia or is a carrier. Identification of mild forms of the disease is important as well meaning physicians may prescribe vitamins or food supplements containing additional iron. While this is appropriate for iron deficiency anemia, it can cause iron overload in children with any form of thalassemia. There is now a simple test for iron in the body called the ferritin level. If someone seems to be iron deficient, but the ferritin level is normal, they probably have the first type of beta thalassemia. Ask your physician to test your child's ferritin level if they are anemic before prescribing vitamins. Thalassemia is a disease that produces abnormal blood cells. You don't need to be a doctor to see that thalassemia cells are NOT NORMAL.
© Copyright 2002 Dr. Nancy Ferguson
Nancy Ferguson is physician (MD) who is board certified in Emergency Medicine, Family Practice and Forensic Medicine. She practicse in an urban hospital with a diverse ethnic patient population having spent three years as a National Health Service Corps volunteer in Appalachia before entering Emergency Medicine full time. She enjoys foreign travel and has lived in Ireland as well as the USA. Her travels with Heart to Heart International have allowed her to lecture at medical schools and hospitals in Armenia, Tanzania, China, Tibet and Costa Rica. She also own a medical legal consulting business. |
||||

|
COMEUNITY http://www.comeunity.com